
平成20年10月21日
水溶性珪素ウモの細菌を用いる復帰突然変異試験
試験期間:2008年9月8日~2008年 10月21日
試験委託者:A・P・Aコーポレーション
試験の経緯: A・P・Aコーポレーションより依頼のあった水溶性珪素ウモ(UMO)について,細菌を用いる復帰突然変異試験にて,安全性を試験した.
試験結果:予試験,本試験ともに陰性であり,遺伝子の突然変異を誘発するような毒性はないと推測される.なお,詳細は別紙の「最終報告書」の通り.
GLP 陳述
試験の表題: 水溶性珪素ウモの細菌を用いる復帰突然変異試験
上記試験は,厚生省令第21号「医薬品の安全性に関する非臨床試験の実施の基準に関する省令」(平成9年3月26日,一部改正厚生労働省令第114号 平成20年6月13 日)に従って実施したものであります.ただし,第13条に記載されている被験物質の特性及び安定性の測定並びに被験物質と媒体との混合物の安定性及び均一性の測定は実施致しておりません.
2008年10月21日
最終報告書
水溶性珪素ウモの細菌を用いる復帰突然変異試験
試験期間: 2008年9月3日~2008年10月21日
2008年10月21日
1. 要約
この試験は,株式会社生物学応用研究所からの委託により実施された.水溶性珪素ウモの遺伝子突然変異誘発性について細菌を用いて検討した.塩基対置換型の遺伝子突然変異を検出するSalmonella typhimurium TA100, TA1535 及びEscherichia coli WP2uvrAの3菌株と, フレームシフト型の遺伝子突然変異を検出するSalmonella typhimurium TA98及びTA1537の2菌株を使用し,プレインキュベーション法により試験を実施した.濃度設定試験及び本試験とも代謝活性化系存在下(以下,代謝活性化法と略)及び非存在下(以下,直接法と略)の条件で試験を実施した.水溶性珪素ウモの用量は,濃度設定試験では,直接法及び代謝活性化法とも公比3で7~5000μg/plateの7用量,本試験では,直接法及び代謝活性化法とも公比2で156.3~5000μg/plateの6用量で実施した.
その結果,濃度設定試験及び本試験とも,いずれの菌株においても陰性対照群コロニー数に対して被験物質群では,用量依存性のある2倍以上の増加はみられず再現性があった.以上の結果から,本実験条件下では,用いた5菌株に対して水溶性珪素ウモに遺伝子突然変異誘発作用はないものと判定された.
2. 試験目的
水溶性珪素ウモの遺伝子突然変異誘発性について細菌を用いて検討した.
3. 試験分担責任者
被験物質液の調製: 山口 裕子
復帰突然変異試験: 今村 匡志
4. 試験期間
試験開始日: 2008年9月3日
濃度設定試験:
前培養; 2008年9月8日
被験物質液添加日; 2008年9月9日
プレート観察日; 2008年9月11日
本試験:
前培養; 2008年9月23日
被験物質液添加日; 2008年9月24日
プレート観察日; 2008年9月26日
試験終了日: 2008年10月21日
5. 遵守する基準及び準拠するガイドライン
GLP: 厚生省令第21号「医薬品の安全性に関する非臨床試験の実施の基準に関する省令」(平成9年3月26日,一部改正 厚生労働省令第114号 平成20年6月13日)
ただし,第13条に記載されている被験物質の特性及び安定性の測定並びに被験物質と媒体との混合物の安定性及び均一性の測定は実施せず.
ガイドライン: 「医薬品の遺伝毒性試験に関するガイドラインについて」(1999年11月1日付医薬審第1604号)
6. 材料及び方法
6.1 被験物質
名称,略称又はコード番号: 水溶性珪素ウモ(以下, UMOと略)
提供元: 試験委託者
ロット番号: 20080818(2008年8月18日製造)
特性:
内容; 水溶性珪素を含んだミネラル複合飲料
性状: 透明(まれにミネラル成分が析出)/アルカリ性
保存条件: 遮光,気密,室温 (条件: 1~30℃,実測値; 16.1~21.9℃)
保存場所: 被験物質保管室
取扱い上の注意: 作業着,マスク及び手袋を着用して取り扱った.
残余分の処置: 試験委託者に返却した.
6.2 対照物質
6.2.1 陰性対照物質(媒体)
名称,略称又はコード番号: 日本薬局方注射用水(以下,注射用水と略)
製造元: 株式会社大塚製薬工場
ロット番号: 8B73P
保存条件: 気密,室温(条件1~30℃)
保存場所: 被験物質保管室
取扱い上の注意: 作業着,マスク及び手袋を着用して取り扱った.
残余分の処置: 未使用分はなかった.
6.2.2 陽性対照物質
名称: 1)Sodium azide (試薬特級,以下NaN3と略)
2)2-(2-Furyl)-3-(5-nitro-2-furyl)acrylamide (和光特級,以下AF-2
と略)
3)2-Aminoanthracene (以下2AAと略)
4) 9-Aminoacridine hydrochloride(以下9AAと略)
製造元: 1),2)及び3)和光純薬工業株式会社
4) MP Biomedicals, LLC
ロット番号: 1) SDK4796
2) WKK3086
3) EWL1809
4)2436F
試験番号:VQ08270
含量・純度: 1)99.8%
2) 99.6%
3) 96.4%
4) 98.8%
原体の保存条件: 1), 2)及び 3); 遮光,室温
4)遮光,冷蔵
原体の保存場所: 1), 2) 及び 3); 被験物質保管室
4)被験物質保管室の保冷庫
調製方法: 1); 注射用水 (ロット番号7K86P)に溶解して後述
する規定濃度に調製した後,フィルター(ポア
サイズ 0.22μm)で遠過した,試験には,凍結
保存したものを解凍して使用した.
2),3)及び4); ジメチルスルホキシド(ロット番号 DPH0023,
和光純薬工業株式会社)にそれぞれ溶解して後
述する規定濃度に調製した.試験には,凍結
保存したものを解凍して使用した.
調製液の保存条件: 遮光,凍結(条件: -80℃以下)
調製液の保存場所: 第2フリーザー室の超低温フリーザー
取扱い上の注意: 作業着,マスク及び手袋を着用し,吸入及び皮膚への付着を防止した.
添加後残液の処置: 業者へ処理を依頼した.
陽性対照物質選択理由: 「医薬品の遺伝癖性試験に関するガイドラインについて」を参考にして選択した.
6.3 添加液
6.3.1 被験物質液の調製
調製時期: 用時調製
調製方法: 濃度設定試験;
1) UMOの 0.2500gに注射用水を加えメスシリンダーを用いて5mLにメスアップした(50 mg/mL).
2) 1)に注射用水を加えて順次希釈し16.67,5.56, 1.85, 0.62,0.21及び 0.07 mg/mL濃度液を調製した(公比3).
本試験;
1) UMOの 0.3001 gに注射用水を加えメスシリンダーを用いて6mLにメスアップした (50 mg/mL).
2) 1)に注射用水を加えて順次希釈し 25, 12.5, 6.25, 3.125及び1.563 mg/mL濃度液を調製した(公比2).
なお,液度設定試験及び本試験ともに,調製の際,ボルテックスミキサーを用いて均一になるまで攪拌した.添加後残液の処理: 業者へ処理を依頼した.
6.4 指標菌株
塩基対置換型: Salmonella typhimurium TA100及びTA1535株(以下,TA100及びTA1535)
Escherichia coli WP2uvrA株(以下, WP2uvrA)フレームシフト型: Salmonella typhimurium TA98及びTA1537株(以下,TA98及びTA1537)
入手先: 国立医薬品食品衛生研究所 変異遺伝部; TA100, TA1535,TA98, TA1537独立行政法人製品評価技術基盤機構; WP2uvrA
入手日: 2004年9月9日 (TA100, TA1535, TA1537)
2005年1月20日 (TA98)
2004年1月29日 (WP2uvrA)
特性検査日: 2007年12月17日~12月20日 (TA100, TA1535, TA98, TA1537及びWP2uvrA)
保存条件: 凍結(条件;-80℃以下)
保存場所: 第2フリーザー室の超低温フリーザー
凍結方法: DMSOを最終強度 8%含む菌懸濁液を調製し,凍結した(1 mL
凍結チュープに200μL).
取扱い上の注意: 用時に菌株を解凍した.
残余分の処置: 解凍後の残余分は,オートクレーブで滅菌後,業者へ処理を依頼した.
試験系選択の理由: 「医薬品の遺伝密性試験に関するガイドラインについて」を参考にして選択した.
6.5 培地
6.5.1 指標菌株の前培養培地(液体栄養培地)
名称: ニュートリエントプロス(NUTRIENT BROTH No.2)
製造元: Oxoid Ltd.
ロット番号: 349915
調製方法: NUTRIENT BROTH NO. 2を蒸留水に溶解して2.5 w/v%とし,L字型試験管に10mLずつ分注した.それらを121℃で15分間高圧蒸気滅菌を行い,使用時まで冷蔵(条件; 1~8℃)した.
使用期限: 調製日を含め1週間
6.5.2 最少グルコース寒天平板培地
名称: クリメディア AM-N培地
原料名: 寒天(伊那寒天 BA-30A,伊那食品工業株式会社)
製造元: オリエンタル酵母工業株式会社
ロット番号: ANI390GX
保存条件: 遮光,室温(条件: 1~30℃)
試験番号:VQ08270
保存場所: 第1遺伝毒性実験室
寒天平板培地の識別: 試験番号,プレート番号を表示するとともに使用する菌がわかるように色ラベルで職別した.
6.5.3 重層用培地
名称: 軟寒天培地(トップアガー)
調製方法: 塩化ナトリウム 0.5 w/v%及び粉末寒天0.6 w/v%を含む軟寒天
培地を121℃で15分間高圧蒸気滅菌した.使用時に溶解し,
S. typhimuriumの4菌株に用いるものには0.5 mmol/Lヒスチジ
ン及び0.5mmol/Lビオチンの混合溶液[フィルター(ポアサイ
ズ0.22μm)で濾過]を,また, E. coli WP2uvrA株に用いるも
のには0.5 mmol/Lトリプトファン溶液[フィルター(ポアサイ
ズ0.22μm)で濾過]をそれぞれ10:1の比で加え,試験操作終
了時まで約45℃で保温した.
保存条件: 0.5 mmol/Lヒスチジン-0.5 mmol/L ビオチン溶液,並びに 0.5
mmol/Lトリプトファン溶液は,冷蔵(条件: 1~8℃).
保存場所: 0.5 mmol/L ヒスチジン-0.5 mmol/L ビオチン溶液,並びに0.5
mmol/Lトリプトファン溶液は,第1遺伝毒性実験室の保冷庫
6.6 薬物代謝活性化酵素系 (S9 mix)
名称: S9 mix (S9及びCofactor-Iの混合物)
Phenobarbital及び5,6-Benzotlavoneで前処理した Sprague-
Dawley 系 7週齢の雄ラットから調製した肝ホモジネート
9000xg の上清画分 (以下,S9と略)に蒸留水に溶解した
Cofactor-I(微生物を用いる変異原性試験用補酵素)を加えた
溶液
ロット番号: 1)S9; 08061303
2) Cofactor-I; 999704
保存条件: 1)S9; 凍結(条件:-80℃以下)
2) Cofactor-I; 冷蔵(条件1~8℃)
保存場所: 1)S9; 第2フリーザー室の超低温フリーザー
2) Cofactor-I; 第1遺伝毒性実験室の保冷庫
調製方法: Cofactor-I バイアルに注射用水(ロット番号 8B73P)を加えて9
mLとしフィルター(ポアサイズ 0.45μm)で濾過した後,この溶液9 mLに対して解凍したS9を1 mLの割合で加えて混合した.調製後は使用時まで氷水中で保存した.
提供源: オリエンタル酵母工業株式会社
組成: S9 mix 1 mL中の組織は, S9 0.1mL, MgCl28μmol, KC133μmol,
G-6-P 5μmol, NADPH 4μmol, NADH 4μmol及びNa-リン酸
緩衝液(pH 7.4) 100μmol.
試験番号: VQ08270
6.7 試験方法
6.7.1 前培養
試験の際には,-80℃以下で凍結保存していた菌懸濁液を解凍し,この液20μLをL字型試験管に分注したNUTRIENT BROTH NO.2の2.5 w/v%液 10 mLに添加した後,振盪培養恒温槽(タイテック株式会社:形式;Personal-11)を用いて振盪培養したものを指標菌液とした.振盪培養条件は,37℃で濃度設定試験は8時間45分,本試験は8時間57分,60回/分1)の速度とした.また,各指標菌液の生菌濃度を濁度計 (富士工業株式会社: A.D.S富士デジタル濁度計)により確認した,生菌濃度は,濃度設定試験では約1.4~3.1×109個/mL,本試験では約1.3~4.1×109個/mLであった.使用時まで菌液を氷水中に保存した.
6.7.2 復帰突然変異試験
試験はプレインキュベーション法2)を用いて,直接法と代謝活性化法について実施した.滅菌試験管に陰性対照物質溶液(注射用水) 0.1 mL,被験物質液 0.1 mL又は陽性対照物質溶液 0.1 mLを入れた.これらに直接法では 0.1 mol/L Na-リン酸緩衝液(pH 7.4) 0.5 mLを,代謝活性化法ではS9 mix 0.5 mLを加え,さらに前培養した指標菌液 0.1 mLを加えて混合し,被験物質などの析出の有無を確認し,37℃で20分間振盪(振盪速度100回/分) しながらプレインキュベートした.次いで,約45℃に保温したトップアガー2 mLを添加混和して速やかに最少グルコース寒天平板培地上に重層し,プレートを裏返して 37℃の恒温槽中で 48時間以上培養した.濃度設定試験及び本試験は試験方法ごとに1群のプレート数は2枚とした.
6.7.3 無菌試験
無菌試験はプレート法により実施した.滅菌試験管に陰性対照物質溶液(注射用水) 0.1 mL,被験物質液 0.1 mL, 0.1 mol/L Na-リン酸緩衝液(pH 7.4) 0.5 mL又はS9 mix 0.5 mLを採取し,トップアガー2 mLを添加混和して速やかに最少グルコース寒天平板培地に重層し, プレートを裏返して 37℃の恒温槽中で48 時間以上培接した.なお,プレート数は各群1枚とした.
6.8 添加量の設定
6.8.1 濃度設定試験
「医薬品の遺伝毒性試験に関するガイドラインについて」を参考に,復帰突然変異試験の最高用量を5000μg/plateとし,以下,公比3で1667,556, 185, 62, 21及び7μg/plateの計7用量を設定した.
陽性対照物質の用量は文献2)を参考に設定した.
被験物質及び陽性対照物質添加群に加え,陰性対照群として媒体(注射用水)を添加する1群を設けた.さらに,無菌試験群を設定し,濃度設定試験に使用した被験物質液,陰性対照群の添加液(注射用水), 0.1 mol/L Na-リン酸緩衝液(pH 7.4)及びS9 mixへの雑菌の混入の有無を確認した.
各指標菌株の陽性対照群については,以下のとおり設定した.
試験番号: VQ08270
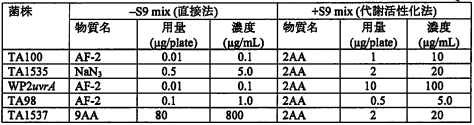
添加液温はすべて 0.1 mL/plateとした.
6.8.2 本試験
濃度設定試験の結果から,本試験用量を以下のように設定した.
濃度設定試験の結果,直接法及び代謝活性化法ともいずれの菌株においても被験物質の析出及び生育阻害はみられなかった.また,陰性対照群復帰変異コロニー数の2倍以上の増加はみられなかった.よって,本試験では生育阻害がみられない4用量以上の評価ができるように,直接法及び代謝活性化法のいずれの菌株も5000μg/plateを最高用量に,以下公比2で2500, 1250, 625, 312.5及び156.3μg/plateの計6用量を設定した.陽性対照群,陰性対照群及び無菌試験群については,「濃度設定試験」の項と同様に設定した.
6.9 観察及び測定方法
1) 各プレートについて,被験物質の試験菌株に対する生育阻害の有無を倒立顕微鏡で観察し,被験物質の析出の有無を肉眼的に観察した.
2) 復帰突然変異により各プレート上に生じたコロニーを,コロニーカウンター(ヤマト科学:形式;CC-21)を用いて計数した.
3) 無菌試験群について,プレート上に菌が生育しているか否かを肉眼的に観察した.
6.10 統計解析
データの統計解析は行わなかった.
6.11 結果の表示及び判定
6.11.1 結果の表示
各プレートの復帰変異コロニー数及び各濃度2枚におけるコロニー数の平均値を表示した.また,被験物質添加時と復帰変異コロニー数計測時の被験物質などの析出及び生育阻害も表示した.
6.11.2 結果の判定
6.11.2.1 陰性及び陽性対照群
陰性対照群の復帰変異コロニー数は,背景データの平均値±3S.D.の範囲内であること,陽性対照群では陰性対照群復帰変異コロニー数の2倍以上の値を示していることを確認した.
試験番号:VQ08270
6.11.2.2 被験物質群
被験物質群における復帰変異コロニー数が,陰性対照群の2倍以上の増加を示し,用量依存性が認められ,かつ濃度設定試験及び本試験の間に再現性が認められた場合に陽性と判定した.陰性対照群の2倍以上の増加がみられない再現性のある結果が得られた場合は陰性と判定した.
6.12 試験成立の基準
以下の基準に該当した場合は,試験は不成立となり再試験を実施することとした.
ない場合.
今回の試験では,上記の基準に該当するものはなかった.
6.13 試験計画書からの逸脱及び予見することができなかった事態
以下のような事例があったが,試験評価に影響を及ぼすものではなかった.
1) 陽性対照物質について,ロット番号 SDJ4376, 含量98.3%のAF-2を使用するよう規定されていたが,実際はロット番号 WKK3086, 含量99.6%のAF-2を使用した,規格は試験計画書規定の和光特級合格品であることから,試験評価に影響を及ぼすものではないと判断した.
6.14 試験関係資料の保存
保存対象: 試験計画書(試験計画変更書含む),最終報告書草案,最終報
告書,生データ及び試験に関連する文書
保存期間: 最終報告書提出後5年間,その後の処置については,所定の
保存期間終了時までに試験委託者と協議する.
地)における所定保管庫
7. 成績
7.1 濃度設定試験
(Table 1)
直接法及び代謝活性化法とも,すべての菌株において被験物質の析出及び菌の生育阻害はみられなかった.また,すべての菌株で陰性対照群復帰変異コロニー数の2倍以上の増加はみられなかった.陽性対照群では,代謝活性化の有無に関わらずいずれの菌株においても陽性反応を示した. 濃度設定試験の結果から,最高用量を5000μg/plateとして,以下公比2で2500, 1250, 625, 312.5及び156.3μg/plate の6用量で本試験を実施した.
7.2 本試験
(Table 2)
直接法及び代謝活性化法とも,すべての菌株において被験物質の析出及び歯の生育阻害はみられなかった.また,すべての菌株で陰性対照群復帰変異コロニー数の2倍以上の増加はみられなかった.陽性対照群では,代謝活性化の有無に関わらず,いずれの菌株においても陽性反応を示した.
7.3 無菌試験
濃度設定試験及び本試験とも,陰性対照物質溶液,被験物質液, 0.1mol/L Na-リン酸緩衝液(pH 7.4)及び S9 mixのいずれにおいても,コロニーの生育は認められなかった.
8. 考察及び結論
濃度設定試験及び本試験とも,陰性対照群の復帰変異コロニー数は当試験実施施設の背景値(2007年7月~2008年8月, Table 3)の平均値±3S.D.の範囲内であった.被験物質群の評価できる用量は4用量以上であった.陽性対照群では,すべての菌株で陽性反応が認められた.また,無菌試験では雑菌の混入は認められなかった.以上の結果から,本試験は成立し,かつ成徴は評価できるものと考えられた.被験物質群では,代謝活性化の有無に関わらず濃度設定試験及び本試験とも,被験物質の析出や菌に対する生育阻害は認められなかった.また,すべての菌株に対する復帰変異コロニー数は,陰性対照群復帰変異コロニー数に対して被験物質群では用量依存性のある2倍以上の増加はみられず,陰性と判定され再現性のある結果であった.以上の結果から,本実験条件下では,用いた5菌株に対してUMOに遺伝子突然変異誘発作用はないものと判定された.
9. 文献
1) 分析機関社内資料.
2) 三宅幸雄, 森田 健, 若田明裕, 朝波省吾, 島田弘康, 日本製薬工業協会 医薬品評価
委員会 基礎研究部会 第3分科会 遺伝毒性ワーキンググループ編集.:医薬品のための
遺伝語性試験 Q&A. 東京: 株式会社 サイエンティスト社; 2000.
VQ08270
Table 1 Bacterial reverse mutation test of Water Solubility Silica UMO (Preliminary test)

a)Negative control, Water for injection b)2-(2-Furyl)-3-(5-nitro-2-furyl)acrylamide
c) Sodium azide d)9-Aminoacridine hydrochloride e)2-Aminoanthracene
VQ08270
Table 2 Bacterial reverse mutation test of Water Solubility Silica UMO (Main test)

a)Negative control, Water for injection b) 2-12-Furyl)-3-(5-nitro-2-furyl)acrylamide
c) Sodium azide d) 9-Aminoacridine hydrochloride e) 2-Aminoanthracene
VQ08270
Table 3 Bacterial reverse mutation test
– Historical background data (2007.7-2008.8).
<Negative control >

信頼性保証部門陳述書
試験の表題 :水溶性珪素ウモの細菌を用いる復帰突然変異試験
試験番号 :VQ08270
上記試験は,下記事項を除き,厚生省令第 21 号「医薬品の安全性に関する非臨床試験の実施の基準に関する省令」(1997年3月26日,一部改正 厚生労働省令第 114号 2008年6月 13 日)に従い,試験計画書及び標準操作手順書に則って行われたことを保証します.
除外事項:被験物質の特性及び安定性の測定並びに被験物質と媒体との混合物の安定性及び均一性の測定
また,最終報告塔には試験の実施方法が正確に記載され,かつ,生データが正確に反映されていることを確認しました.
信頼性保証部門による調査は「信頼性保証部門陳述書(別紙)」に示す日程で行われています.なお,「信頼性保証部門陳述書(別紙)」プロセス調査記載の調査項目については定期調査の結果をもって本試験の操作の保証としました.
添付資料:信頼性保証部門陳述書(別紙)2枚
本陳述書は添付資料を含め全3枚
2008年10月31日
試験調査
運営管理者への報告日
試験計画書 2008年9月18日 2008年9月22日 兼松ひろみ
試験計画変更書-1 2008年9月22日 2008年9月22日 兼松ひろみ
細菌を用いる復帰突然変異試験(試験操作) 2008年9月23日及び24日 2008年10月6日 兼松ひろみ
被験物質,媒体の保管 2008年9月24日 2008年10月6日 兼松ひろみ
被験物質の調製及び使用 2008年9月24日 2008年10月6日 兼松ひろみ
細菌を用いる復帰突然変異試験(観察) 2008年9月26日及び10月2日 2008年10月6日 兼松ひろみ
試験記録 2008年10月14日及び15日 2008年10月15日 兼松ひろみ
報告書草案(図表) 2008年10月14日及び15日 2008年10月15日 兼松ひろみ
報告書草案(本文) 2008年10月14日及び15日 2008年10月15日 兼松ひろみ
試験記録【再調査】 2008年10月21日 2008年10月21日 兼松ひろみ
報告書草案(本文)【再調査】 2008年10月21日 2008年10月21日 兼松ひろみ
報告書最終案(図表) 2008年10月21日 2008年10月21日 兼松ひろみ
報告書最終案(本文) 2008年10月21日 2008年10月21日 兼松ひろみ
最終報告書 2008年10月21日 2008年10月21日 兼松ひろみ
信頼性保証部門陳述書(別紙)
プロセス調査
調査項目 調査日 運営管理者への報告日 調査実施者
被験物質の受領,返却及びサンプル保存 2008年7月29日及び30日 2008年7月30日 中島 隆
細胞の性状確認及び継代・菌株の特性試験 2008年7月22日~25日 2008年7月28日 兼松ひろみ
及び保存